Research
ApolloX: a Conditional Modeling for Amorphous Multi-Element Materials

In modeling amorphous materials—One of the most structurally complex systems in condensed matter—the principle of less is more becomes essential. Unlike crystals, whose periodicity simplifies structure representation, amorphous solids lack long-range order, making brute-force enumeration or exhaustive sampling infeasible. Instead, progress relies on identifying minimal yet physically meaningful structural descriptors that capture local order, guide energy landscape exploration, and enable efficient prediction. In this context, simplicity is not a compromise, but a necessity—where fewer, well-chosen parameters can unlock deeper understanding of disorder and glass formation. arXiv:2503.07043 [cond-mat.mtrl-sci]
Data-Driven Design of Battery Electrolytes and Electrode Materials
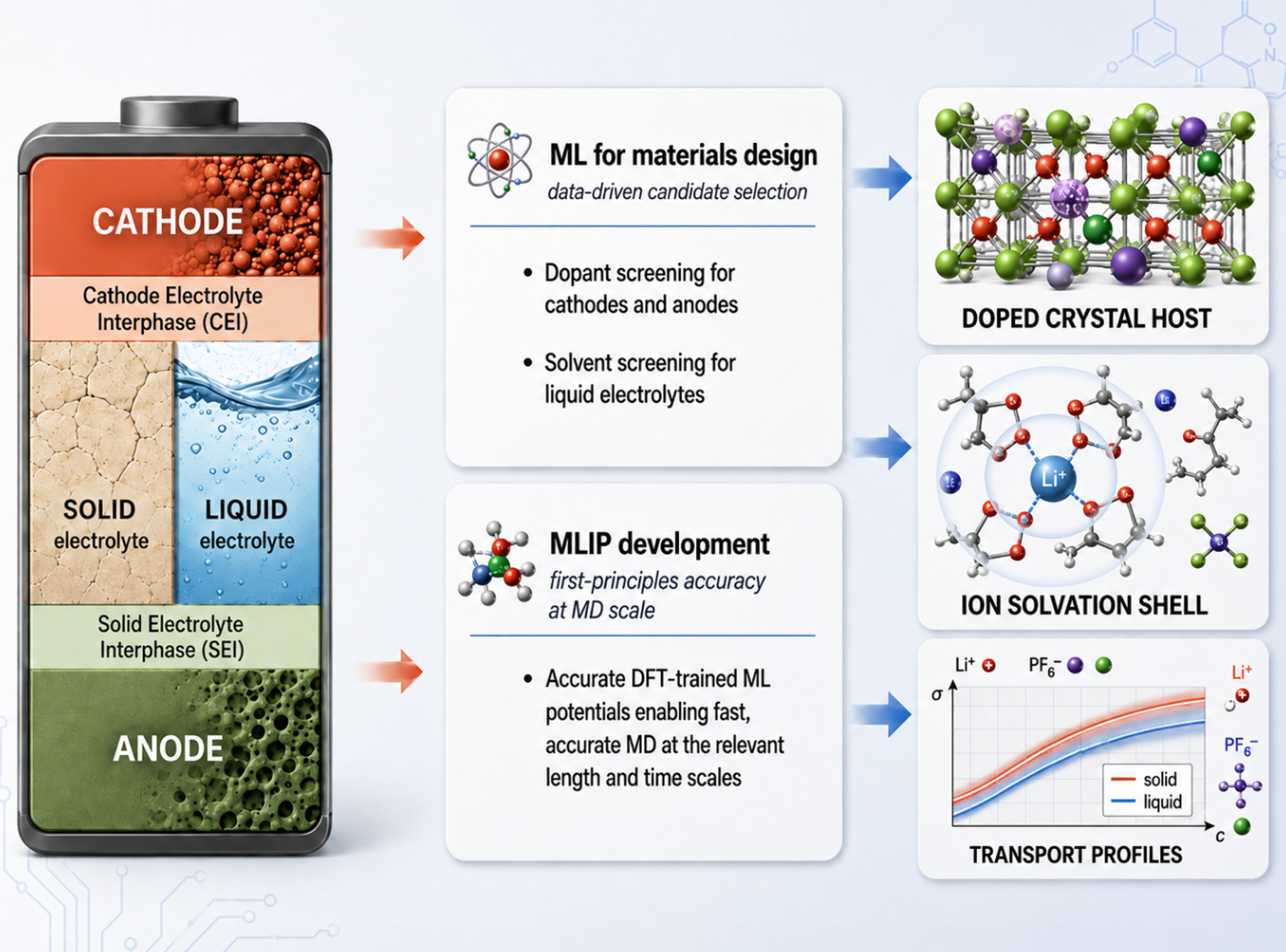
Our research leverages machine learning to accelerate the discovery and understanding of next-generation battery materials, with a focus on both solid and liquid electrolyte systems. On the materials design front, we develop data-driven screening frameworks to identify promising dopants for cathode and anode hosts, as well as to evaluate candidate solvents for liquid electrolyte formulations. Complementing this effort, we build machine learning interatomic potentials (MLIPs) trained on first-principles (DFT) data, enabling molecular dynamics simulations that retain quantum-mechanical accuracy while reaching the length and time scales required to capture realistic battery chemistry. Together, these approaches allow us to probe doped crystal hosts, ion solvation structures, and ionic transport profiles across diverse electrolyte environments, providing atomistic insight into the interfacial and bulk phenomena that govern battery performance, stability, and safety.
Advancing Computational Modeling and Design for Catalysts under Realistic Conditions

Computational modeling plays a pivotal role in unraveling the intricate mechanisms and enhancing the efficiency of catalysts. However, comprehensively discerning the local environment surrounding a catalytic center under authentic conditions, encompassing surface engineering, thermalization, solution effects, electric potentials, catalyst dynamics, revolutions, and the operando characterization of structural changes during catalytic reactions, remains a formidable challenge. Our research group is dedicated to modeling real conditions for a diverse array of catalysts, encompassing cluster-based heterogeneous catalysts, enzymatic catalysts, and supramolecular capsules, especially focus on the reactions of water splitting, CO2 reduction and N2 reduction. This profound understanding of the real nature of the active sites is paramount in the design of high-performance catalysts.
Electronic Structure, Spectroscopy and Chemical Bonding of Size-Selected Metal (3d/4f/5f) Nanoclusters and Nanoalloys

Nanoclusters offer valuable and systematic means to investigate the electronic structure, chemical bonding, and catalytic mechanisms of catalysts and materials. The evolving geometries and increasing sizes of clusters provide insights into their growth mechanisms under different experimental conditions, including confinement on specific surfaces. Within our research group, one of our focal projects involves exploring transition metal, lanthanide, and actinide clusters. Our aim is to unravel intriguing chemical bonding phenomena and spectroscopic characteristics (such as vibrational, electronic, and dynamics of excited states). These investigations lay a theoretical foundation for catalyst design and hold promise for developing novel catalytic materials.
Design of Boron-Based Self-Assembly Nanomaterials

The recent discovery of two-dimensional boron materials, known as “borophene,” similar to carbon’s “graphene,” has led to increased interest in exploring new boron-based materials. Boron clusters, which have received less attention than carbon clusters, present unique challenges due to boron’s electron deficiency and the diverse structures that arise as the clusters grow. Highly symmetrical clusters often serve as building blocks for material design. Therefore, studying the stability, geometry revolution, electronic structure, and chemical bonding of metal-doped boron clusters, particularly those with lanthanide and actinide elements, is crucial. This research can unlock the potential of boron-based materials with exceptional magnetic, optical, and catalytic properties, leading to significant technological advancements.
Machine Learning of Atomic Potential for Study of Descriptor for Materials and Catalyst Design

In this specialized area, our research group is dedicated to harnessing the capabilities of machine learning to advance our comprehension of atomic potentials, encompassing energy and force. By seamlessly integrating these potentials into force fields with chemical accuracy, we can conduct multiscale calculations, employing techniques like ML/MM (machine learning/molecular mechanics), for molecular dynamics or metadynamics simulations across extensive time and length scales. Moreover, we are committed to identifying informative fingerprint descriptors, leveraging insights from quantum chemistry and molecular dynamics, which prove instrumental in designing catalysts with enhanced stability and selectivity. The amalgamation of machine learning techniques and domain-specific expertise holds the promise of expediting the discovery of efficient catalysts and materials, endowed with tailored properties.
Supported by:




